
Wat verandert er door nieuwe Europese regels en welke maatregelen moeten zorginstellingen nemen?
Inleiding
Ziekenhuizen willen steeds vaker ‘stille Intensive Care-afdelingen’ (IC’s). Door akoestische alarmen van de aanwezige medische apparatuur te onderdrukken, heeft de patiënt meer rust en dit versnelt het herstel [1,2]. Naast stille IC’s zien we ook meer éénpersoonskamers in medium en low care-afdelingen. Informatie over de patiënt en vooral over de alarmen zullen in dat geval met hoge betrouwbaarheid doorgezet moeten worden naar de zorgverleners, die op eenduidige wijze alarmen en berichten willen ontvangen. Net als verpleegoproepen, moeten belangrijke medische alarmen direct naar de dienstdoende zorgverleners worden doorgezet. Ze houden hierdoor beter overzicht en kunnen efficiënter zorg verlenen. De hoeveelheid aan alarmen die verpleegkundigen ontvangen, kan in ziekenhuizen heel groot zijn. Verpleegkundigen zouden alleen de relevante alarmen moeten ontvangen zodat alarmmoeheid (wat in de top 10 van gevaren in de zorg staat) vermindert.
De vraag naar een Medisch Oproep Systeem (MOS) groeit door deze ontwikkelingen. Een modern MOS kan alarmen bundelen en prioriteren en ondersteunt daarmee zowel patiëntveiligheid als efficiënte inzet van de verpleegkundige capaciteit. Een MOS valt echter onder de certificering van medisch hulpmiddel in de Medical Device Regulatory (MDR), oftewel de Verordening betreffende Medische Apparatuur [3].
De MDR, officieel de Verordening (EU) 2017/745, is in mei 2017 gepubliceerd en in werking getreden. Elke fabrikant, leverancier en zorginstelling moet na de transitieperiode van 3 jaar, de wet vanaf 25 mei 2020 toepassen. De regulering vervangt de huidige European Medical Devices Directive (MDD) en definieert veiligheids- en effectiviteitseisen voor alle medische apparatuur die wordt verkocht in de Europese Unie, en kwaliteitsgaranties voor fabrikanten, importeurs en distributeurs.
Onder de nieuwe MDR valt ook gedegen onderzoek en een risicoanalyse bij de aanschaf van hulpmiddelen, waaronder systemen voor alarmdistributie. Maar wat zegt deze regulering nou precies, wat is er veranderd en hoe kan de nieuwe regelgeving worden benut om patiëntveiligheid te verbeteren?
Medische hulpmiddelen
Elk medisch hulpmiddel dat in Europa op de markt wordt gebracht of ‘in huis’ wordt vervaardigd, valt onder de MDR. Een medisch hulpmiddel is een ‘instrument, apparaat, software of ander artikel dat door de fabrikant is bestemd om te worden gebruikt voor diagnose, preventie, monitoring, behandeling of verlichting van ziekte, letsel of beperking’ bij de mens. Een MOS monitort en is dus een medisch hulpmiddel [4] conform de MDR. Indien er ook patiëntdata wordt meegestuurd en het alarm inhoud heeft die kan worden gebruikt voor diagnose en preventie, valt het dus ook onder de MDR.
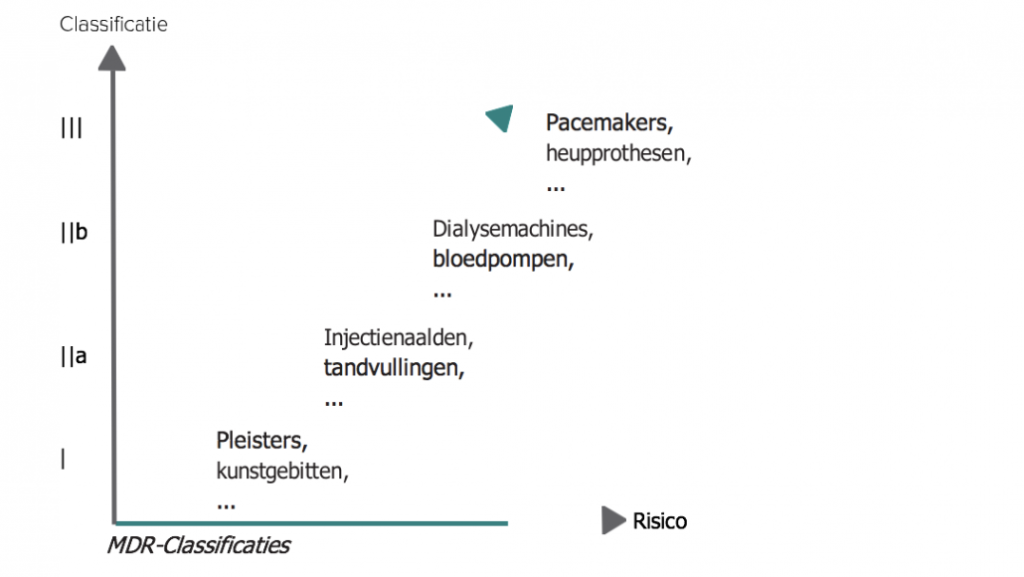
Om recht te doen aan de grote verscheidenheid in toepassingen en risico’s van medische hulpmiddelen, kent de MDR vier classificaties met oplopend risicoprofiel en daarmee gepaard gaande vereisten.
Daarnaast kent de MDR het begrip ‘toebehoren van een medisch hulpmiddel’. Dit zijn artikelen die door de fabrikant bestemd zijn om samen met medische hulpmiddelen te worden gebruikt om bij te dragen aan de medische functionaliteit van de medische hulpmiddelen. Ook op het toebehoren zijn de classificatieregels van toepassing. De MDR onderkent ook het begrip ‘systeem’: dat is een combinatie van producten die al dan niet samen zijn verpakt en bestemd zijn om onderling te worden gekoppeld of te worden gecombineerd om een specifiek medisch doeleind te bereiken.
MDR vervangt MDD
De MDR is vastgesteld om inherente lacunes in de oude Medical Device Directive (MDD) te adresseren. Daarnaast moet de MDR ervoor zorgen dat de regelgeving aansluiting houdt bij de snelle evolutie van wetenschap en technologie op het gebied van medische apparatuur en hulpmiddelen.
Deze nieuwe regulering heeft drie doelgebieden: veiliger aankopen, veiliger implementeren en veiliger gebruiken. Om die doelen te halen, zijn er diverse methoden en maatregelen voorgeschreven.
3.1 Belangrijke wijzigingen ten opzichte van de MDD
De MDR is niet de nieuwe versie van de MDD. De MDR is allereerst een verordening, in plaats van een richtlijn (zoals de MDD). Een verordening heeft directe werking binnen de EU. Oftewel; alle lidstaten van de EU zijn gebonden aan dezelfde regels, omdat sprake is van een ‘directe werking’. Daarnaast omvat de MDR een aantal belangrijke aanscherpingen ten opzichte van deze eerdere regelgeving.
Enkele belangrijke wijzigingen zijn:
- Een strenger controlemechanisme voor ‘hoge risico’-apparatuur en -hulpmiddelen via een strenger toezicht vooraf door experts op Europees niveau.
- Strengere criteria voor aanwijzing van en processen voor toezicht op zogeheten notified bodies.
- De instelling van een uitgebreide EU-database over medische hulpmiddelen en een traceerbaarheidssysteem op basis van unieke apparaat-identificatie.
- De versterking van de regels inzake klinisch bewijs, met inbegrip van een EU-wijde gecoördineerde procedure voor toelating van klinische onderzoeken.
- De versterking van toezicht vereisten na het in de handel brengen van fabrikanten.
- Verbeterde coördinatiemechanismen tussen EU-landen op het gebied van waakzaamheid en markttoezicht.
Daarnaast kent de MDR nieuwe regels voor software, waarvan de belangrijkste is dat software, die een hulpmiddel bestuurt of het gebruik ervan beïnvloedt, in dezelfde klasse valt als het betreffende hulpmiddel.
Ten opzichte van de MDD is het begrip ‘intended use’ oftewel beoogd gebruik ongewijzigd. Dit wordt door de fabrikant vooraf vastgelegd en bepaalt het doel waarvoor een zorginstelling het hulpmiddel, apparaat of systeem mag inzetten om binnen de grenzen van de MDR te blijven.
3.2 Classificatie van medische alarmering binnen de MDR
Een systeem voor medische alarmering valt dus onder de MDR. De classificatie ervan vindt plaats op basis van het door de leverancier beschreven beoogd gebruik. Daarbij is het risico voor de patiënt leidend.
Actieve hulpmiddelen voor diagnostische en monitoring- doeleinden behoren tot klasse IIa, indien zij bestemd zijn om een directe diagnose of monitoring mogelijk te maken van vitale fysiologische functies.
Als de aard van de variaties van die parameters echter zodanig is dat deze tot onmiddellijk gevaar voor de patiënt kunnen leiden, of zij bestemd zijn voor diagnose in klinische situaties waarin de patiënt in onmiddellijk gevaar verkeert, dan behoren zij tot klasse IIb. Denk hierbij bijvoorbeeld aan variaties in de prestaties van het hart, de ademhaling en de activiteit van het centrale zenuwstelsel.
3.3 Classificatie van software binnen de MDR
De definitie van software is in de MDR veranderd ten opzichte van de MDD. Het is daarom belangrijk om aan de hand van de nieuwe regels te bepalen of uw software valt onder de MDR. Door de nieuwe classificatieregels valt meer software die bestemd is voor therapeutische en diagnostische doeleinden in een hogere risicoklasse dan voorheen. Als deze software de basis is voor een besluit dat het overlijden van een patiënt of een onomkeerbare verslechtering van zijn gezondheidstoestand tot gevolg kan hebben, valt deze volgens de nieuwe regels zelfs in de hoogste risicoklasse: klasse III. Meer informatie over de classificatie van software staat in onderstaande tabel.
 Doelgebieden MDR
Doelgebieden MDR

Impact van MDR op zorgorganisaties
De gevolgen van de strengere regelgeving zijn allereerst voelbaar bij fabrikanten van medische hulpmiddelen. Zij zullen de hulpmiddelen conform de MDR moeten ontwerpen, produceren en laten certificeren. Toch moet de impact op zorgorganisaties niet worden onderschat.
Certificering
De zorgorganisaties moeten allereerst voor alle vanaf 2020 op de markt aangeboden hulpmiddelen van hun leveranciers nieuwe certificeringen, inclusief beoogd gebruik, opvragen. Tegelijkertijd moeten ze valideren dat het -al dan niet veranderde- beoogd gebruik past bij hun gebruik van het hulpmiddel. Daarnaast gaat de MDR uit van een medewerking van de zorginstelling bij ‘post market surveillance’ activiteiten door de zorgautoriteit en/of notified body.
Traceerbaarheid
Er komt een unieke identificatiecode op alle medische hulpmiddelen te staan. Deze identificatie zorgt voor betere traceerbaarheid van hulpmiddelen. Zorginstellingen en patiënten kunnen zo eerder worden geïnformeerd als er een gebrek aan een hulpmiddel is geconstateerd. De nieuwe regels stellen echter wel dat zorginstellingen moeten zorgdragen voor het bewaren van de unieke identificatiecode. Dit geldt vooralsnog alleen voor implanteerbare medische hulpmiddelen met een hoog risico. In de toekomst kunnen hier ook andere risicocategorieën aan worden toegevoegd.
Zelf samenstellen
Zorginstellingen produceren zelf ook medische hulpmiddelen. Soms uit noodzaak, omdat marktpartijen (nog) geen passende oplossing hebben en soms uit wetenschappelijke interesse. De MDR stelt dat vanaf mei 2020 zorginstellingen die zelf hulpmiddelen vervaardigen (juridisch gezien) als fabrikant gekwalificeerd worden, inclusief alle bijkomende verantwoordelijkheden en verplichtingen uit de MDR. In dat geval dient de zorginstelling er tenminste voor in te staan dat aan elk van de volgende voorwaarden wordt voldaan:
- de hulpmiddelen worden niet overgedragen aan een ander rechtspersoon;
- de hulpmiddelen worden vervaardigd en gebruikt met inachtneming van een passend kwaliteitsmanagementsysteem 5 ;
- de zorginstelling rechtvaardigt in haar documentatie dat aan de specifieke behoeften van de
- patiënten-doelgroep niet kan worden voldaan, of daaraan niet op een passend prestatieniveau kan worden voldaan, door een op de markt beschikbaar gelijkwaardig hulpmiddel;
- de zorginstelling verstrekt haar bevoegde autoriteit op verzoek informatie over het gebruik van bedoelde hulpmiddelen, waaronder een rechtvaardiging voor de vervaardiging, de wijziging en het gebruik ervan;
- de zorginstelling stelt een verklaring op, die ze openbaar maakt en de naam en adres van de vervaardigende zorginstelling bevat, gegevens ter identificatie van de hulpmiddelen, een verklaring waaruit blijkt dat de hulpmiddelen voldoen aan de algemene veiligheids- en prestatie-eisen;
- de zorginstelling stelt documentatie op met uitleg over de productiefaciliteit en het productieproces, het ontwerp en de prestatiegegevens van de hulpmiddelen, met inbegrip van het beoogde doeleind, die voldoende gedetailleerd is om de bevoegde autoriteit in staat te stellen te beoordelen of er wordt voldaan aan de algemene veiligheids- en prestatie-eisen;
- de zorginstelling neemt alle maatregelen die nodig zijn om te garanderen dat alle hulpmiddelen in overeenstemming met de onder 6) bedoelde documentatie worden vervaardigd;
- de zorginstelling evalueert de ervaring die is opgedaan met het klinisch gebruik van de hulpmiddelen en onderneemt alle vereiste corrigerende acties.
Om aan de nieuwe wetgeving te voldoen zullen veel zorginstellingen op het gebied van zelf produceren van medische hulpmiddelen en systemen dan ook een steile leercurve door moeten maken.
Integraal of zelf integreren?
Het includeren van software, strengere classificaties en zwaardere eisen aan vigilantie die de MDR stelt, zijn daarom aspecten om terdege rekening mee te houden bij ontwerp en inzet van uw MOS. Daarnaast is vanuit bedrijfseconomisch oogpunt een geïntegreerde oplossing gewenst die zo veilig mogelijk is, die eenvoudig te beheren is en die het primaire zorgproces naadloos ondersteunt met de benodigde garanties.
DIS, DAS, CDAS
Bij het beschrijven van alarmeringssystemen worden vaak de termen DIS, DAS en CDAS gebruikt. Deze termen komen uit IEC 80001-2-5:2014 6 . Hierin worden de risico’s beschreven van IT-systemen waarin medische apparatuur is opgenomen. Het is dan ook van belang de verschillen te onderkennen tussen deze varianten.
Van oudsher gebruiken zorginstellingen technologie voor het distribueren van alarmen naar pagers of (Wifi/DECT) handsets van zorgverleners. Aangezien er altijd risico’s zijn met het gebruik van technologie, zoals systeemuitval, kan het systeem zelf niet garanderen dat de alarmen altijd bij het juiste personeel terechtkomen. Deze beperking zorgt er voor dat een dergelijk systeem alleen gebruikt wordt ter notificatie en niet voor besluitvorming. Hierdoor classificeert een dergelijk systeem zich als een gedistribueerd informatie systeem (DIS) in plaats van een alarmeringssysteem.
Om een gedistribueerd alarmeringssysteem (DAS) te realiseren dient er vanaf de handset een technische bevestiging gestuurd te kunnen worden naar het medisch apparaat dat het alarm gegenereerd heeft. Krijgt het medisch apparaat geen bevestiging, dan kan het nogmaals het alarm verzenden en/of escaleren. Hierdoor wordt het risico dat een alarm door een technisch falen niet aankomt, geminimaliseerd. Er kan dan echter nog steeds niet met zekerheid worden vastgesteld of het alarm is ontvangen en geaccepteerd door de zorgverlener. Daarvoor dient een laatste stap gezet te worden: de bevestiging van de zorgverlener vragen en terugkoppelen in het systeem. Hiermee wordt een gedistribueerd alarmeringssysteem met operator confirmation (CDAS) gecreëerd.
Alleen met een DAS en CDAS wordt de keten voldoende betrouwbaar om als klasse IIb alarmeringssysteem ingezet te kunnen worden. En kan een grote stap gezet worden naar stille IC’s en veilige éénpersoonskamers. Het samenstellen van een DAS of CDAS vraagt daarom om samenwerking van vele partijen. Zowel binnen de zorginstelling (denk aan de medische instrumentatie dienst, de klinisch fysici, de ICT-afdeling, de facilitair manager, zorgmanagers) als buiten de instelling.
Conclusie
Een succesvolle implementatie van een DAS of CDAS binnen de kaders van de MDR, die alle voordelen optimaal benut en meetbare klinische voordelen biedt, zal daarom interdisciplinair opgezet moeten zijn en vanuit één centraal verantwoordelijke moeten worden aangestuurd. Een werkwijze die bij de introductie van Elektronische Patiënten Dossiers (EPD’s) in Nederland zijn nut reeds heeft aangetoond. Zo zullen voor alle betrokken afdelingen de voordelen in het dagelijks werk merkbaar worden. Hierdoor zal de oplossing de hele keten optimaal ondersteunen.
Daarbij staan zorginstellingen wel voor een belangrijke tactische keuze: óf de losse bouwstenen aanschaffen óf kiezen voor één integraal systeem. Deze keuze is cruciaal voor de organisatie. Wordt het DAS of CDAS door de zorginstelling zelf samengesteld uit bouwstenen van meerdere leveranciers (en wellicht wat eigen geproduceerde bouwstenen) dan ligt de klinische validatie, certificering en ‘post market surveillance’ ook bij de instelling. Wordt het systeem als geheel bij een consortium of integrator aangeschaft, dan kan daar een belangrijk deel van de taken worden belegd.
Zo kan een op maat gemaakte, modulaire oplossing van verschillende vendoren binnen de regelgeving gerealiseerd worden. En als de leverancier/integrator, zoals Ascom, al internationale samenwerking heeft, waardoor interoperabiliteit vooraf al getoetst is tussen de verschillende vendoren, kan dit de invoering versnellen. Waarbij naast medische alarmering ook andere workflows eenvoudiger ondersteund kunnen worden.
Een aanpak waarbij vanuit één verantwoordelijkheid binnen het ziekenhuis met één leverancier wordt samengewerkt om het medisch alarmeringssysteem vorm te geven, lijkt dan ook voor veel ziekenhuizen en zorginstellingen de meeste kansen te scheppen om zich tijdig en zonder onnodige kosten aan de snel veranderende regelgeving aan te passen. Door de interoperabiliteit tussen bestaande medische hulpmiddelen, zorginformatiesystemen en managementoplossingen, wordt de informatie over de zorg ook inzichtelijker, toegankelijker en beter bruikbaar. Hierdoor zijn snellere reactietijd, meer tijd voor zorg en hogere patiëntveiligheid een stap dichterbij.




{kind=link}